Project goal: To assess the role of Src kinases in the CSF-1R signaling pathway
Recent literature implicates a regulatory function of the juxtamembrane domain (JMD) in receptor tyrosine kinases. Mutations in the JMD of c-Kit and Flt3 are associated with gastrointestinal stromal tumors and acute myeloid leukemias, respectively. Additionally, autophosphorylated Y559 in the JMD of the Colony Stimulating Factor-1 (CSF-1) receptor (CSF-1R) binds to Src kinases.
The SH2 domain of the p85 subunit of PI3-kinase (PI3K) binds to phosphorylated Tyr721 in the kinase insert (KI) of CSF-1R. A mutant CSF-1R deleting the entire KI (DKI) does not bind p85 and lacks receptor-associated PI3K activity. Surprisingly, DKI-CSF-1R retained the ability to activate downstream effectors of PI3K, Akt and p70s6 kinase, in a PI3K-dependent manner. We hypothesize that PI3K activation is mediated by Src kinases which bind phosphorylated Tyr 559 in CSF-1R. This hypothesis is supported by our demonstration that Src kinases are activated by CSF-1 in 32D premyeloid cells expressing either wildtype (WT) or DKI CSF-1R and by our finding that Akt activation is sensitive to a specific chemical inhibitor of Src kinases and to expression of a dominant-negative Src.
Another level of complexity has been added by the recent demonstration that juxtamembrane phosphorylatable tyrosines, such as Tyr 559 may be involved in autoinhibitory regulation (Fig. 1). We have made Src binding mutants of the CSF-1R (Y559F, Y559D, I562S) to assess the role of Src binding in PI3K activation and to determine if Tyr 559 has a dual function, that of providing docking sites for Src and of autoregulation. These mutants have been expressed in 32D myeloid cells by retroviral transduction. The cell lines have been used to assess the importance of Src in mediating various CSF-1-activated signaling pathways.
While binding to I562S was not significantly perturbed, Y559F and Y559D exhibited markedly decreased CSF-1-dependent SFK association. All JMD mutants retained intrinsic kinase activity, but Y559F, and less so Y559D, showed dramatically reduced CSF-1-induced autophosphorylation (Fig. 2). CSF-1-mediated wild-type (WT)-CSF-1R phosphorylation was not markedly affected by SFK inhibition, indicating that lack of SFK binding is not responsible for diminished Y559F phosphorylation. Unexpectedly, cells expressing Y559F were hyperproliferative in response to CSF-1 (Fig. 3). Hyperproliferation correlated with prolonged activation of Akt, Erk and Stat5 in the Y559F mutant. Consistent with a defect in receptor negative regulation, c-Cbl tyrosine phosphorylation and CSF-1R/c-Cbl co-association were almost undetectable in the Y559F mutant. Further, Y559F underwent reduced multiubiquitination and delayed receptor internalization and degradation (Fig. 4).
In conclusion, we propose that Y559 is a switch residue that functions in kinase regulation, signal transduction and, indirectly, receptor downregulation. These findings may have implications for the oncogenic conversion of c-Kit and Flt3 with JMD mutations.
We are extending our studies on the Src family kinases (SFKs) to bone marrow-derived macrophages using a combination of approaches: progenitors from knockout mice and retroviral-mediated silencing or overexpression. We are specifically interested in how different SFKs individually and in combination regulate macrophage proliferation and differentiation.
See our latest publication on the role of Src kinases pdf.
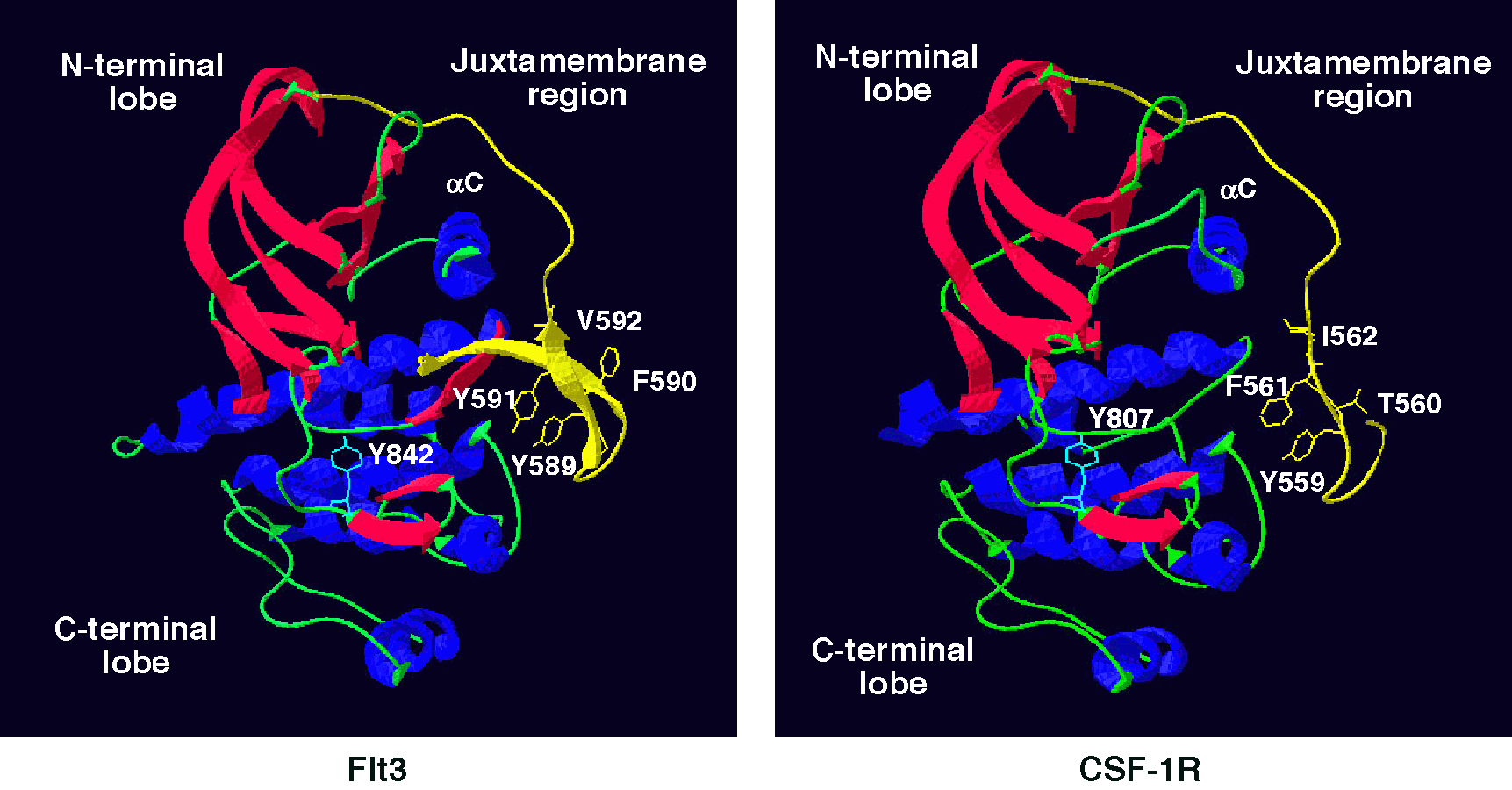
Fig. 1. Model for the Juxtamembrane domain of the CSF-1R: comparison to the crystal structure of Flt3.
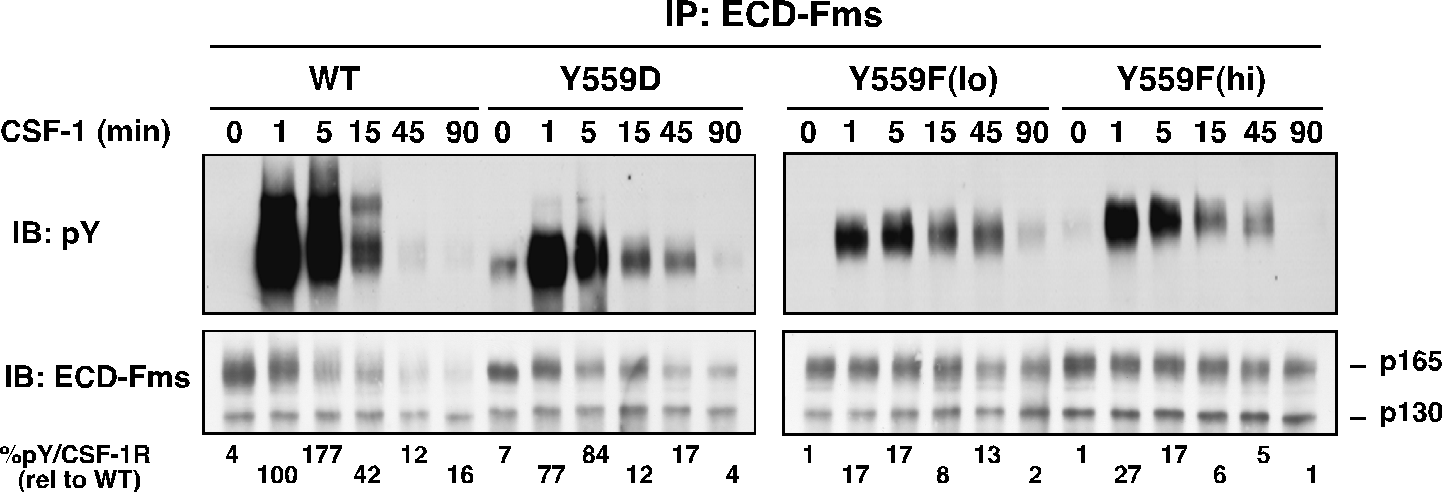
Fig. 2. Receptor autophosphorylation is much diminished in juxtamembrane CSF-1R mutants.
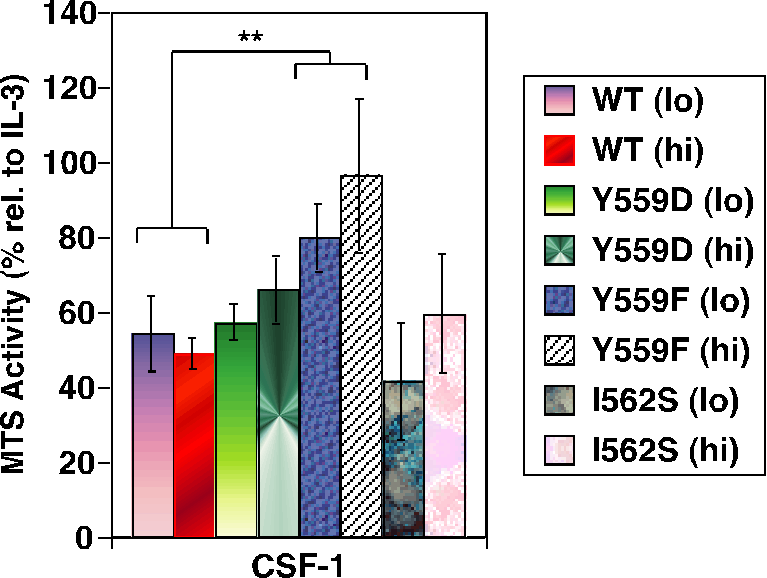
Fig. 3. Juxtamembrane CSF-1R mutants hyperproliferate in response to CSF-1.
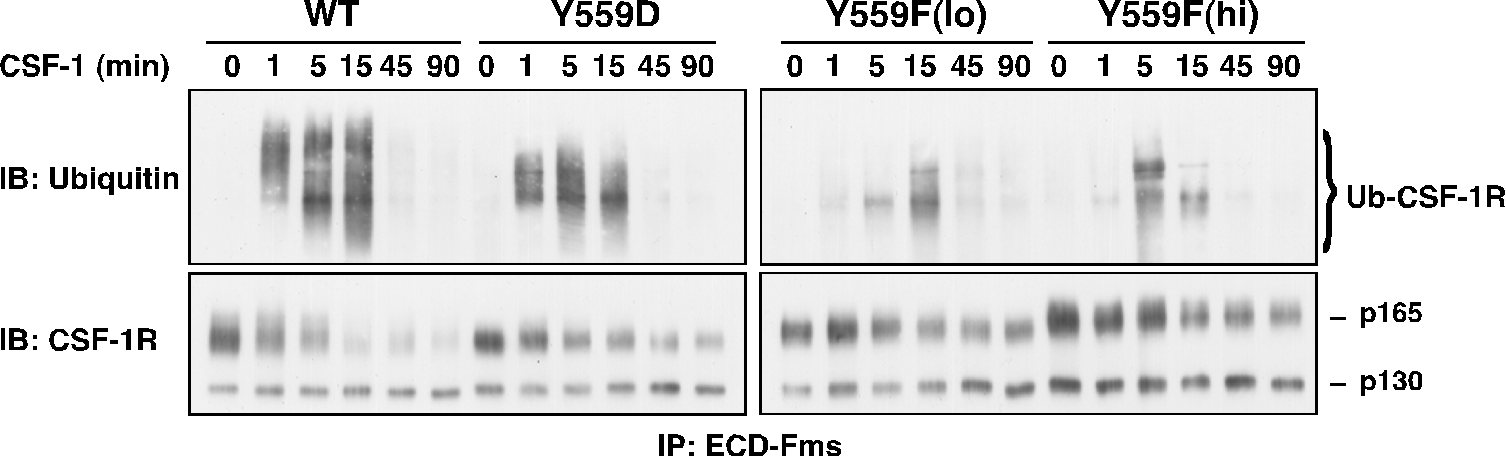
Fig. 4. Juxtamembrane CSF-1R mutants show diminished cbl-mediated ubiquitination.